Congenital pulmonary airway malformation (CPAM) is a benign tumor of the lung characterized by a cystic mass of disorganized but otherwise normal lung tissue. It is thought to occur from abnormal development of the embryonic lung bud at the fifth or sixth week of development. CPAMs can occur in any lobe of the lung and rarely affects more than 1 lobe. CPAM is slightly more common in males and rarely presents before 20 weeks of gestation.
CPAMs are usually diagnosed on routine prenatal ultrasound, but rarely before 20 weeks gestation. After 20 weeks gestation, the CPAM grows very rapidly. There is a classification system that divides CPAM into 5 categories:
- Type 0 represents 1 to 3% of cases in which small cysts cover almost all of the lungs, and this type is incompatible with life.
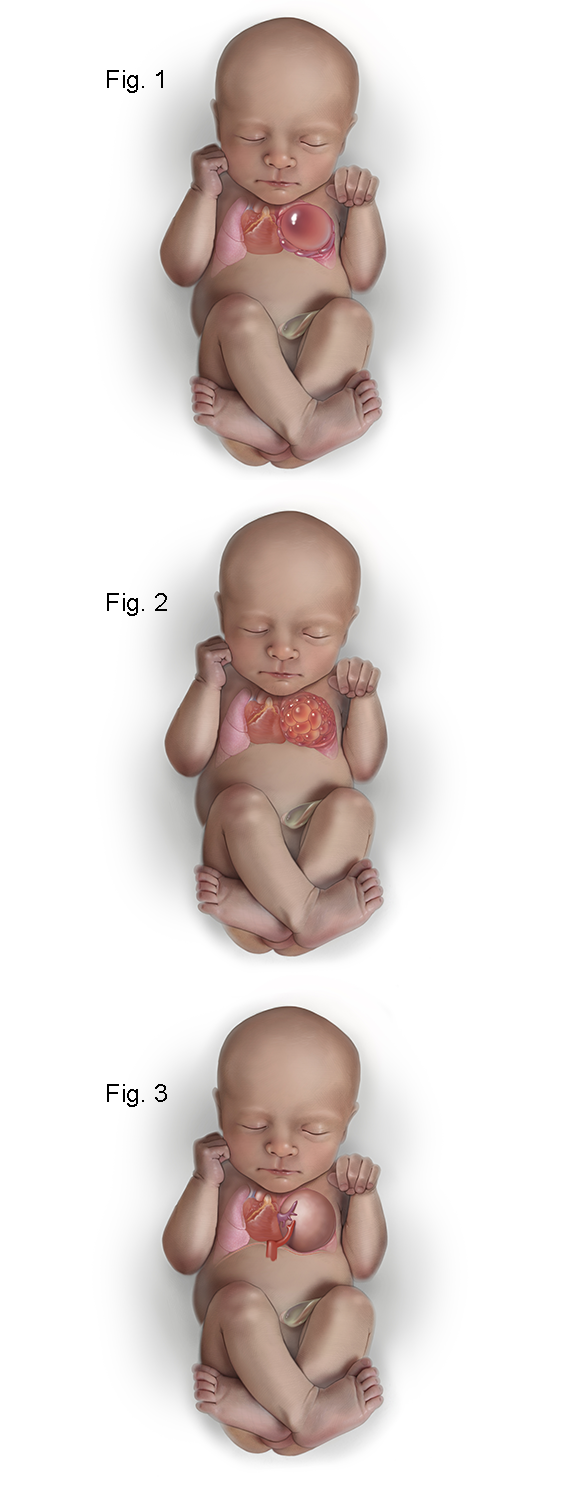
- Type I CPAMs account for 50% of cases and have relatively few large cysts, ranging in size from 3 to 10 cm. Figure 1 shows the fetus with a left upper lobe type I congenital pulmonary airway malformation that has a large unilocular cyst which is shifting the heart and mediastinum to the left and compressing the left lower lobe.
- Type II CPAMs account for 40% of cases and have numerous cysts of smaller size, usually less than 1 cm. Type II CPAMs are associated with multiple congenital anomalies. Figure 2 shows the fetus with a type II CPAM in which the cysts are smaller than in type I.
- Type III CPAMs are usually large homogeneous, solid-appearing masses, which can grow to large sizes in utero and result in nonimmune hydrops. Figure 3 shows a type III CPAM which is microcystic and on ultrasound appears solid. In addition, there is a systemic arterial feeding vessel making this a “hybrid” CPAM which has features of both a CPAM and a bronchopulmonary sequestration (BPS).
- Type IV CPAMs account for 15% of cases and are characterized by very large cysts, up to 10 cm in size.
It is important to distinguish low-risk and high-risk CPAMs based on the CVR. CPAMs with CVR < 1.6 rarely get into trouble and only need to be followed on a weekly basis until a plateau is reached at approximately 26 weeks gestation. In contrast, a CPAM with a CVR > 1.6 is at markedly increased risk for the development of hydrops. In these cases, we treat mothers with two intramuscular injections of betamethasone, a steroid that helps arrest the rapid growth of the CPAM. In high-risk CPAMs with a CVR > 1.6 and no hydrops, treatment with prenatal steroids has been associated with 100% postnatal survival. In high-risk CPAMs with CVR > 1.6 and hydrops, steroids may still be effective with 40% postnatal survival. In addition, these hydropic CPAMs may respond to additional courses of betamethasone if hydrops do not resolve after the first course.
In cases unresponsive to betamethasone, fetal surgery may be indicated. Cases with a large cyst may benefit from thoracoamniotic shunt placement. In some CPAM cases, fetal thoracoscopy may be used to create communications between all the cysts within the CPAM to maximize the effect of the thoracoamniotic shunt that is used to decompress it (see Figures 4-7 below).
Figure 4. The illustration shows the fetoscope has been inserted into the fetal chest and into the dominant cyst of the CPAM with a laser fiber (green tip) extended at the end of the fetoscope for laser fenestration of non-communicating cysts in the CPAM.
Figure 5. The illustrations shows a close up of the laser fiber extended from the tip of the fetoscope in order to create a communication with the dominant cyst of non-communicating cyst by using laser energy to burn a hole in the cyst wall.
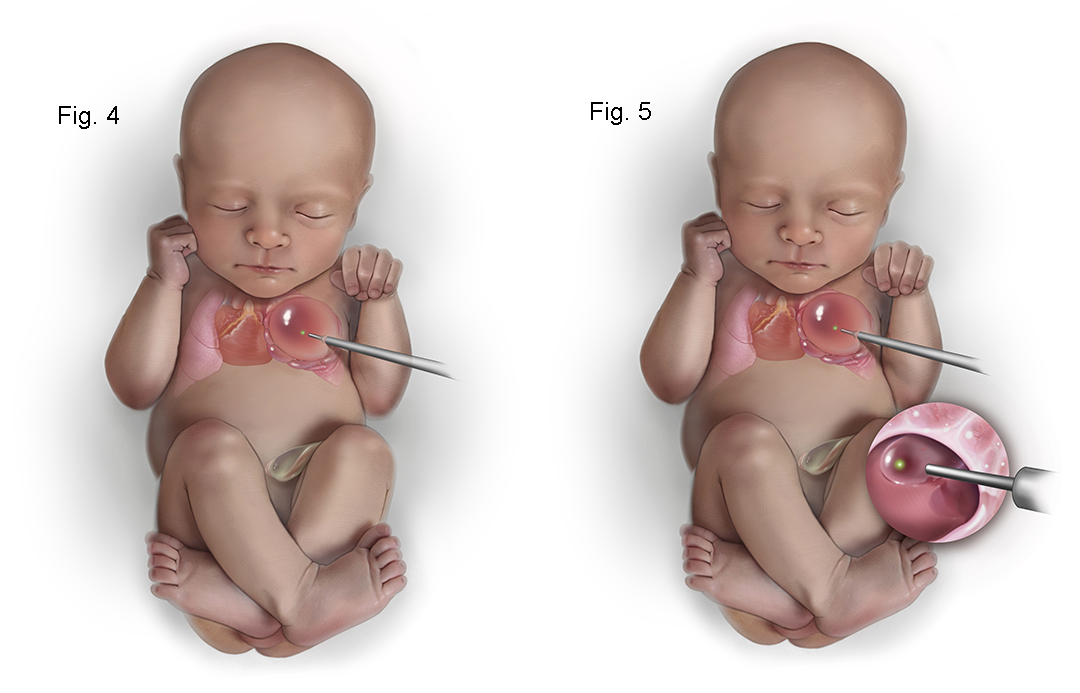
Figure 6. The illustration shows a close up view of the internal CPAM showing communication of the cysts with the dominant cyst allowing more complete decompression of the CPAM.
Figure 7. The illustration shows the placement of a thoracoamniotic shunt with a double pigtail catheter which allows drainage of fluid within the CPAM to the amniotic cavity. Internally the shunt sits with a pigtail within the CPAM and it passes through the chest wall with a pigtail on the outside to drain fluid into the amniotic cavity.
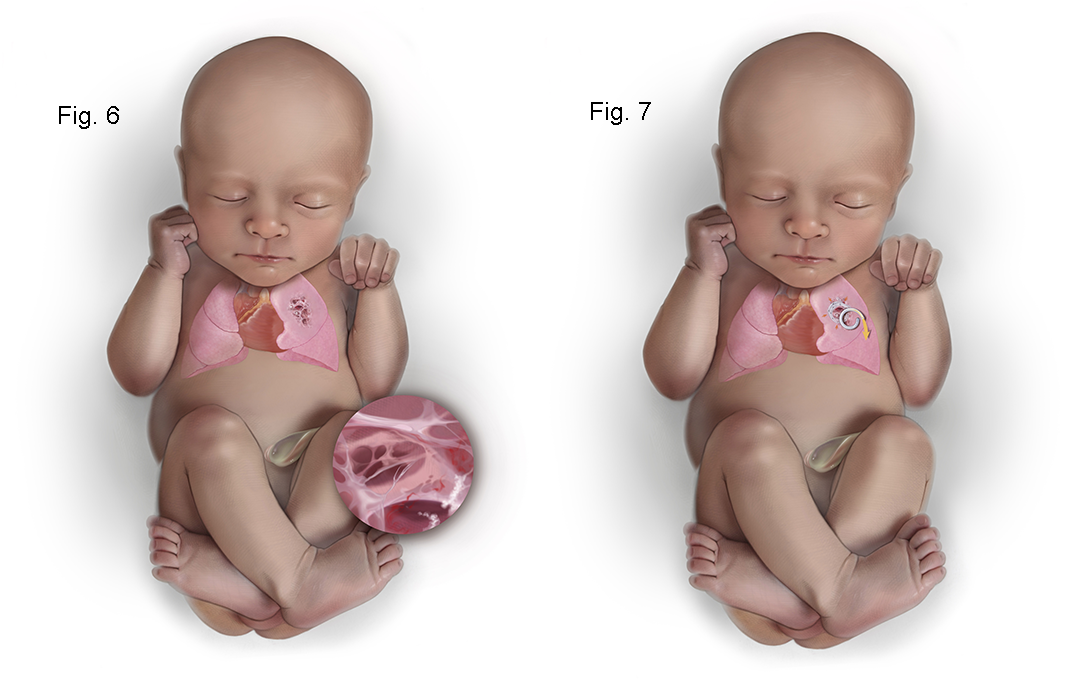
In solid type III CPAMs with hydrops, shunting is of no benefit. If unresponsive to steroids, the only option may be open fetal surgery for thoracotomy and lobectomy. This is rarely necessary with the use of steroids, but open fetal surgery has a 65% survival rate. A newer less invasive approach is ultrasound-guided intravascular laser photocoagulation (see Figures 8-9 below). The lobar pulmonary artery to the lobe with a CPAM is coagulated causing regression in the size of the CPAM. This allows the baby to grow around the CPAM. This approach does not sacrifice any lung function because the CPAM provides no functional gas exchange after delivery.
Figure 8. The illustration shows how a type III hybrid CPAM can be treated with intravascular laser photocoagulation. An 18 gauge needle is passed under ultrasound into the CPAM into the systemic feeding vessel and the laser fiber is inserted into the vessel just beyond the tip of the 18 gauge needle. Turing on the laser fiber uses light to coagulate the systemic feeding vessel which prevents the CPAM from growing and causes it to regress in size.
Figure 9. The illustration shows the 18 gauge needle in the systemic feeding vessel with the laser fiber extended into the vessel lumen about to be fired to photocoagulate the vessel.
For Medical Professionals
Congenital cystic adenomatoid malformation (CCAM), or the more recent term congenital pulmonary airway malformation (CPAM) of the lung, is a lesion characterized by a multicystic mass of pulmonary tissue with a proliferation of bronchial structures (1,2). It may represent a failure of maturation of bronchiolar structures, occurring at approximately the 5th or 6th week of gestation, during the embryonic stage of lung development (1-3). Alternatively, it may represent focal pulmonary dysplasia, since skeletal muscle has been identified within the cyst walls (4). Others have suggested that it may be the result of airway obstruction (5-8). The gestational age and location of the airway obstruction may determine whether CPAM, bronchopulmonary sequestration, or lobar emphysema results (9,10).
The normal fetal lungs have a homogeneous appearance with moderately high signal intensity on T2-weighted images (49). The T2 signal intensity increases with gestational age and T1 signal intensity decreases with gestational age. This effect is thought to be secondary to the increasing number of alveoli as gestation progresses as well as increasing lung fluid production (50). In the setting of a CPAM, the compression of the normal lung by the mass of the CPAM may make the adjacent normal lung appear lower in signal intensity on T2 images compared to the contralateral normal lung (49). The liver, mediastinal structures, and lung are easily differentiated from each other by their signal intensity.
Fetal MRI is a valuable adjunct to ultrasound in the evaluation of CPAMs. It is often not possible to determine which lobe is involved in the CPAM by ultrasound. In contrast, fetal MRI is often able to delineate the T2 signal intense CPAM from compressed normal lobes which have a lower T2 signal compared to normal lungs. Fetal MRI has proven useful when the diagnosis based on ultrasound alone is uncertain and increases the specificity of the diagnosis (51-53).
Macrocysts within CPAMs are highly signal intense of T2 weighted images with adjacent solid components of the CPAM also being high on T2-weighted images. CPAMs that are entirely macrocystic are hyperintense on T2. Microcystic CPAMs may be difficult to distinguish from lobar or segmental hyperinflated lung. The presence of cysts confirms the diagnosis of CPAM, but in the absence of macrocysts, it may be difficult to distinguish a hyperinflated lung from a type III CPAM. The vascular distribution in a hyperinflated lobe or segment is normal, whereas in a CPAM, the normal vascular distribution is disrupted by irregular spacing of vessels by signal intense parenchyma. It may not be possible to distinguish microcystic or type III CPAMs from hyperinflation on prenatal MRI and postnatal thin-section CT scans with intravenous contrast may be needed to make this distinction with certainty. It may also be difficult to distinguish a microcystic hybrid CPAM with a systemic arterial feeding vessel from an intralobar bronchopulmonary sequestration. Both BPS and a type III CPAM appear homogeneously signal intense on T2 weighted images and both will have a flow void from the systemic feeding vessel. A concomitant pulmonary arterial feeding vessel can be seen in hybrid CPAMs as well as intralobar BPS.
It is not well established what is the optimal timing in gestation to image CPAMs. Few CPAMs present prior to 20 weeks gestation, and most MRIs are obtained in the late second trimester at the time a diagnosis is made or suspected on prenatal ultrasound. Some have suggested that repeat MRI in late gestation improves diagnostic accuracy and may reduce the need for postnatal imaging. Late third-trimester fetal MRI may be helpful in deciding whether or not an EXIT-to-Resection may be indicated. While there is no consensus about the indications for the EXIT procedure for CPAMs, it is thought that a shift of the mediastinum, causing complete effacement of the intrathoracic trachea, would be an indication for the EXIT procedure (54). This would be difficult to determine by ultrasound in the later third trimester as there is usually less amniotic fluid, and the acoustic windows for imaging the fetal chest are limited.
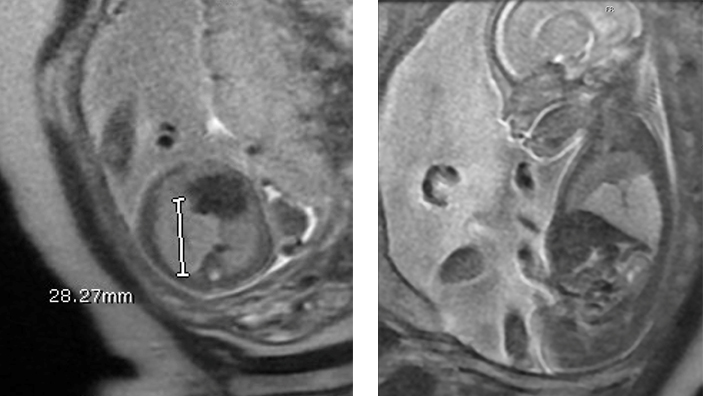
There have been several MRI-derived quantitative measurements reported to predict the risk of developing hydrops and postnatal respiratory compromise. MRI-specific quantitative measurements include the fetal MRI-derived lung mass volume ratio (LMVR) (which is the MRI equivalent of the CVR), the lesion-to-lung volume ratio (LLV), and the observed/expected normal fetal lung volume (O/E-NFLV) (55). Of these MRI-derived measurements, the LMVR was found to be the strongest predictor of the development of hydrops and neonatal respiratory distress after delivery. But there was no direct comparison made between LMVR and CVR. While LMVR is measured by the sum of serial coronal MRI images, CVR is measured by 3-dimensional measurements obtained by ultrasound and normalized for gestational age by dividing by the head circumference. One would predict that LMVR would be equivalent to CVR as they are measuring the same thing using different modalities.
Although in the majority of cases, CPAMs are isolated lesions, in 10-20%, particularly in type II lesions, there can be associated anomalies as noted above (56,57). MRI can be particularly helpful in excluding these anomalies or defining in fetuses known to have a CPAM.
The postnatal natural history of CPAM can be quite variable (36,58). The lesion can be completely asymptomatic and come to medical attention only when chest radiography is performed for other reasons, such as a history of mild respiratory complaints or recurrent infections in infancy or childhood. However, most postnatal patients will present at birth with severe cardiorespiratory compromise due to severe pulmonary hypoplasia (1,33,59-65). Even before the advent of obstetrical sonography, it was recognized that up to 14% of cases of CPAM result in stillbirths (1). This observation hinted at the different prenatal natural history of CPAM.
Our understanding of the natural history of CPAM is still evolving. The worst outcome is observed in fetuses in which hydrops develops (19,20,66). Hydrops is usually seen in very large lesions, often type III, which cause mediastinal shift and vena caval obstruction. Hydrops may also be exacerbated by the loss of protein from the CPAM into the amniotic fluid, thus reducing the fetal colloid oncotic pressure from hypoproteinemia (33). Anecdotal reports exist of fetuses with CPAM surviving after the onset of hydrops (67-75). Diamond et al., suggested that resolution by 30 weeks’ gestation may be more common than is appreciated (76). The reason for this unexpected resolution of hydrops in CPAM was not apparent until the natural history of CPAM was defined by Crombleholme (35). CPAMs plateau in their growth at an average of 26 weeks’ gestation, after which the fetus grows around the CPAM allowing hydrops to resolve (35).
The overall prognosis depends primarily on the size of the lesion, as well as whether it is predominantly macrocystic or microcystic. Polyhydramnios is seen in up to 70% of CPAMs diagnosed antenatally (36). The pathogenesis of polyhydramnios is not completely understood but is thought to relate to esophageal obstruction from mediastinal shift and interference with fetal swallowing of amniotic fluid (2,77). This is supported by the absence of fluid in the fetal stomach in such cases.
The diagnosis of CPAM may also have implications for the health of the mother. The mother with a fetus with CPAM may develop the “mirror syndrome,” a hyperdynamic preeclamptic state that may be life-threatening. The “mirror syndrome” has also been seen in molar pregnancies, sacrococcygeal teratoma, and in fetal conditions that result in poor placental perfusion, which leads to endothelial cell injury (78,79). The only treatment for this syndrome is immediate delivery of the baby and placenta.
The antenatal diagnosis of a large CPAM might at first appear to be an ominous finding; however, several older reports have described disappearing fetal lung masses (58,66,80-83). MacGillivray et al., have reported six cases of large CPAM with associated mediastinal shift that progressively decreased in size over the course of gestation (83). These lesions were all of the microcystic, or type III, variety but none was associated with hydrops. It is now believed that these “disappearing” CPAMs are an artifact of changing echogenicity of the lung at 32-34 in which normal lung becomes isoechogenic with the CPAM. If an MRI is obtained, the CPAM is readily distinguished from normal lung on T2 weighted images despite having “disappeared” by ultrasound.
One of the largest experiences with prenatally diagnosed CPAMs was reported by Adzick et al., who described 134 fetuses with CPAM (36). Of these, 14 pregnancies were terminated, 101 cases were managed expectantly, 13 women underwent open fetal surgery, and 6 fetuses underwent thoracoamniotic shunt placement. In the fetuses that did not develop nonimmune hydrops, the postnatal survival was 100%. In contrast, of 25 large CPAMs that developed hydrops and were managed expectantly, there was 100% mortality, with death in utero or immediately after birth. Among the 76 fetuses with CPAMs that were not associated with hydrops, the uniform survival was, in part, due to planned near-term delivery at a tertiary care center. Many of the babies with large lesions required substantial ventilatory support, and four needed support with extracorporeal membrane oxygenation (ECMO).
Fifteen CPAM lesions appeared large at 20 to 26 weeks of gestation with an associated contralateral mediastinal shift, but then clearly decreased in size during the third trimester with the return of the position of the heart toward midline. Although four of these shrinking lesions were associated with polyhydramnios, including one case with fetal ascites, these phenomena resolved as the masses decreased in size.
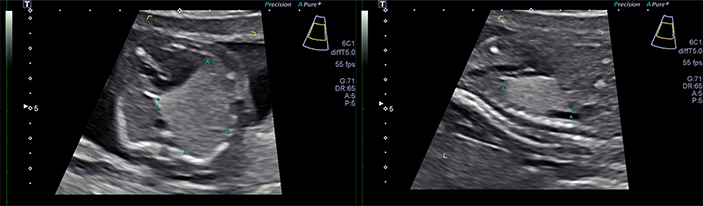
Crombleholme et al., reported the use of CPAM volume and the CPAM volume ratio (CVR) which helped define the prenatal natural history of CPAMs and as a predictor of the development of hydrops (34). The CPAM volume is calculated using the formula for the volume of an ellipse (h × w × l × 0.52 in cm3) with the measurement of the greatest length in the sagittal section and the width and height taken at 90 degrees to the sagittal measurement. The CPAM volume ratio (CVR) is obtained by dividing the CPAM volume by the head circumference (in cm) to correct for any differences in gestational age. Based on 32 fetuses with CPAMs, the CPAM volume and the CVR at presentation were found to be significantly higher in fetuses destined to develop hydrops. The mean CVR plus 2 standard deviations of fetuses that did not develop hydrops yielded a CVR of 1.6 which comprised 95% of CPAMs with a favorable prognosis. Only 2% of fetuses with CVR < 1.6 developed hydrops, and all of these had a dominant cyst. Of those fetuses with CVRs >1.6, 80% developed hydrops. The CVR may therefore be a useful criterion for selecting fetuses at the greatest risk for the development of hydrops and those at low risk for the development of hydrops (34).
The use of the CVR allowed the prenatal natural history of CPAMs to be better defined. CPAMs are rarely diagnosed prior to 20 weeks gestation and then grow rapidly until a mean gestational age of 26 weeks at which time the growth plateaus. After this point, the CVR declines as the baby grows around the CPAM. This behavior is useful in following fetuses with CPAMs as once a growth plateau is reached the CPAM will regress in size.
The use of betamethasone in high-risk CPAMs has become standard practice despite the lack of direct evidence that steroids arrest the growth of CPAMs. In a systematic review of the efficacy of antenatal steroids in the management of high-risk CPAMs, Patwardhan et al. identified 13 observational studies involving 163 pregnancies complicated by CPAM that were treated by single or multiple courses, of antenatal steroids (86). They found a significant mean decrease in the CVR of 1.16 and resolution of hydrops in 86%. Not all patients respond to a single course of steroids, and Morris et al., have reported that two-thirds of patients who do not respond to an initial course of steroids will respond to a second course (87). Fetal surgery for CPAMs is now reserved for only those cases that prove refractory to at least two courses of steroids.
The management of CPAM and hydrops depends on the CVR value that is obtained at presentation. If the CVR is less than 1.6 and there is no evidence of a dominant cyst, the CPAM has little risk for the development of hydrops (34). The fetus should have weekly sonograms to measure the CPAM volume and CVR in order to identify early signs of hydrops or, more likely, that the plateau in growth has been reached. Once the growth plateau is reached, the pregnancy is no longer at risk for the development of hydrops. The surveillance of the fetus can be reduced but one should continue to assess the size of the CPAM, the risk of pulmonary hypoplasia, or “air trapping” that would influence delivery management.
If there is a dominant cyst, even if the CVR is less than 1.6, the fetus remains at significant risk for acute enlargement of the cyst and development of hydrops. A thoracoamniotic shunt may be considered in these cases at the very earliest sign of hydrops.
If the CVR is more than 1.6 at presentation, with or without a dominant cyst, there is up to an 80% chance of hydrops developing (35). Twice weekly sonographic surveillance should be started to help detect the earliest signs of hydrops. Maternal administration of antenatal corticosteroids should be considered in all cases with a CVR of 1.6 at presentation. To date, there have been 13 case series reported with apparent resolution of hydrops in patients with CPAMs that were treated with antenatal steroids (87-89). It is thought that steroids may arrest the growth of the solid component of the CPAM inducing an early growth plateau and allowing the fetus to grow around the CPAM and hydrops to resolve. Due to the lack of a prospective randomized clinical trial, it is not proven that steroids truly affect the growth of CPAMs. It is still possible that the observations reported may simply reflect CPAMs naturally entering a growth plateau regardless of steroid administration.
Fetuses with CPAM and a dominant cyst in which hydrops develop prior to 32 weeks can be considered for treatment in utero. There now have been multiple reports of cystic CPAMs successfully treated by thoracoamniotic shunting (37,38,73,75,90-94) with the survival of 70% of fetuses. Thoracoamniotic shunting for CPAMs with a large cystic component is now considered routine. Many of these cysts do not communicate, however, which may limit the reduction in volume that can be achieved with thoracoamniotic shunt alone. Crombleholme and colleagues have used fetal thoracoscopy to fenestrate the cyst walls of a CPAM to enhance the reduction in volume obtained with thoracoamniotic shunting (see Figures 4-7 below). Typically, up to 50% reduction in CPAM volume may be achieved with thoracoamniotic shunt alone. In contrast, fetal thoracoscopic fenestration of CPAM cysts plus thoracoamniotic shunt can achieve up to 70% reduction in CPAM volume. However, these survivors may still be at risk for associated pulmonary hypoplasia and marked respiratory insufficiency at birth and some have required high-frequency ventilation or even ECMO support.
Figure 4. The illustration shows the fetoscope has been inserted into the fetal chest and into the dominant cyst of the CPAM with a laser fiber (green tip) extended at the end of the fetoscope for laser fenestration of non-communicating cysts in the CPAM.
Figure 5. The illustrations shows a close up of the laser fiber extended from the tip of the fetoscope in order to create a communication with the dominant cyst of non-communicating cyst by using laser energy to burn a hole in the cyst wall.
Figure 6. The illustration shows a close up view of the internal CPAM showing communication of the cysts with the dominant cyst allowing more complete decompression of the CPAM.
Figure 7. The illustration shows the placement of a thoracoamniotic shunt with a double pigtail catheter which allows drainage of fluid within the CPAM to the amniotic cavity. Internally the shunt sits with a pigtail within the CPAM and it passes through the chest wall with a pigtail on the outside to drain fluid into the amniotic cavity.
The most challenging antenatal presentation is large microcystic CPAM with hydrops that does not lend itself to shunt decompression. Fetal surgical resection of massively enlarged microcystic CPAM with associated hydrops has been performed in at least 25 patients at 21 to 29 weeks gestation (58,95). In this series, of the 16 fetuses that survived (64%), CPAM resection led to hydrops resolution in 1 to 2 weeks, return of the mediastinum to the midline within 3 weeks, and impressive in utero lung growth. There were also nine fetal deaths in this series.
The need for open fetal surgery for CPAM has largely disappeared since the advent of steroid use to arrest the growth of these lesions. Open fetal surgery presents significant potential risks for the mother with only ~64% survival for affected fetuses who undergo the procedure. An attractive alternative that is far less invasive than open fetal surgery is ultrasound-guided intravascular laser ablation of the lobar branch of the pulmonary artery supplying the CPAM (96) (see Figures 8-9 below). The CPAM is not functional lung tissue and there is little downside to ablation of the branch pulmonary artery to the CPAM. There is limited experience with treating CPAMs with this approach but more experience in treating BPS (97). However, this approach to devascularize the CPAM appears to be safe involving much less risk for the mother with good expectation for survival of the fetus.
Figure 8. The illustration shows how a type III hybrid CPAM can be treated with intravascular laser photocoagulation. An 18 gauge needle is passed under ultrasound into the CPAM into the systemic feeding vessel and the laser fiber is inserted into the vessel just beyond the tip of the 18 gauge needle. Turing on the laser fiber uses light to coagulate the systemic feeding vessel which prevents the CPAM from growing and causes it to regress in size.
Figure 9. The illustration shows the 18 gauge needle in the systemic feeding vessel with the laser fiber extended into the vessel lumen about to be fired to photocoagulate the vessel.
As mentioned above, a course of maternal steroids (betamethasone or dexamethasone) may be effective in arresting the growth of the CPAM (86-89). In some instances, the size of the CPAM remains substantial with significant mediastinal shift and cardiac compression. In these cases, delivery by EXIT-to-Resection may be indicated (98). The rationale for this approach is that the mediastinal shift and compression by the CPAM will make ventilation difficult and will similarly impair venous return if ECMO is attempted. During EXIT-to-Resection, a thoracostomy for resection of the CPAM is performed on placental support. In this approach when the infant is born the trachea is decompressed facilitating ventilation, and if ECMO is needed, venous return will be unobstructed (98). In a small series of 9 cases of large CPAMs managed by EXIT-to-Resection, Hedrick et al reported 8 of 9 survived and 4 subsequently required ECMO support (98).