Congenital diaphragmatic hernia (CDH) is among the most challenging anomalies to manage in the neonatal intensive care unit. The reason for this difficulty is primarily due to the pulmonary hypertension associated with CDH. We have demonstrated that having a dedicated CDH Team can dramatically improve survival and reduce the need for extracorporeal membrane oxygenation support. The CDH Team is involved in the management of the baby with CDH from the time of prenatal diagnosis, and delivery, throughout the NICU stay, and long-term follow-up. The CDH Team is composed of specialists in Maternal Fetal Medicine, Fetal Surgery, Neonatology, Pediatric Surgery, Fetal Radiology, and Fetal and Pediatric Cardiology.
A mother carrying a baby with CDH, first meets the CDH Team during her initial comprehensive prenatal CDH assessment. This assessment utilizes prognostic variables obtained from fetal MRI, ultrasound, and fetal echocardiography to develop a CDH composite prognostic index or CDH-CPI. The CDH-CPI predicts the severity of the CDH, the severity of associated pulmonary hypertension, and the impact of any associated anomalies on the outcome by integrating multiple prognostic variables to predict anticipated survival, the likelihood of the need for ECMO support, and estimate the length of stay in the NICU.
The diagnosis of CDH is usually made by routine ultrasound when the fetal stomach bubble is noted to be adjacent to the fetal heart in the case of left-sided CDH. Right-sided CDH may be more difficult to recognize as the liver is usually herniated and has an echotexture similar to that of the adjacent fetal lung.
Ultrasound is an important imaging modality not only to diagnose the CDH but also to detect other anomalies which might be present, such as genitourinary, gastrointestinal, abdominal wall, and intracranial abnormalities. Important prognostic criteria are also obtained, including the lung-to-head circumference ratio (LHR) and observed-to-expected LHR. Fetal biometry is also obtained to assess fetal growth and Doppler velocimetry of the umbilical artery, umbilical vein, ductus venosus, and the middle cerebral artery.
Fetal MRI is invaluable in confirming the presence of CDH, determining the presence and degree of liver herniation, and providing multiple lung measurements that provide prognostic information. In CDH, genetic abnormalities are present in up to 20% of cases and have a disproportionate impact on survival and other outcomes. We always recommend that mothers have a genetic amniocentesis for karyotype analysis and microarray analysis. This may also have implications for the mother’s delivery. In cases in which there may be lethal anomalies, urgent cesarean delivery for fetal distress would be contraindicated as it would not benefit the baby and expose the mother to unnecessary risk. Fetal MRI is an essential tool in evaluating a baby with CDH. Fetal MRI provides exquisite anatomic detail of the hernia and better defines the degree of liver herniation. In addition, fetal MRI is used to measure total lung volume (TLV), observed to expected total lung volume (O/E TLV), and percent predicted lung volume (PPLV).
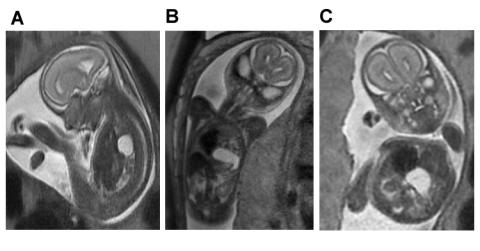
A. Saggital view of severe left-sided CDH demonstrating herniation of the left lobe of the liver filling the anterior chest and displacing the fluid filled stomach posteriorly.
B. Coronal view of the same fetus demonstrating the fluid-filled stomach rotated 180-degrees counterclockwise, sitting in the chest next to the heart.
C. Axial view of the same fetus demonstrating the relative positions of the shifted heart, herniated liver and stomach.
Fetal echocardiography is essential to rule out congenital heart defects, which may be present up to 20% of cases of CDH. In many instances, the presence of congenital heart defects may not alter the overall prognosis, survival, or long-term outcomes. However there are some critical structural heart defects, which may adversely affect outcomes due to the need for intervention to achieve adequate systemic or pulmonary blood flow, for example hypoplastic left heart syndrome. Other forms of congenital heart disease, such as a large unrestrictive ventricular septal defect may compound the risk of pulmonary hypertension. In addition to determining structural heart disease, fetal echocardiography is used to identify left ventricle to right ventricle disproportion as a marker of severity, rule out arrhythmias, and assess the severity of pulmonary hypertension.
We use the McGoon index and the prenatal pulmonary hypertension index (PPHI= the diameter of the left main pulmonary artery (in left CDH) divided by the craniocaudal length of the cerebellar vermis) to assess the severity of pulmonary hypertension. In the McGoon index, the sum of the diameters of the branch pulmonary arteries divided by the aortic diameter at the level of the diaphragm yields a ratio that is prognostically important. A McGoon index of less than 0.9 indicates the baby will have severe pulmonary hypertension and will be have pulmonary pressures equal to or greater than systemic pressures at 3 weeks of age. In contrast, when the McGoon index is greater than 1.2 the baby is likely to have less severe pulmonary hypertension with pulmonary pressure is significantly less than systemic pressure by 3 weeks of age. In the PPHI, a ratio of < 1.2 indicates the baby will have severe pulmonary hypertension with pulmonary pressures equal to or exceeding systemic pressures at 3 weeks of age. In contrast, a PPHI of > 1.5 indicates the baby’s pulmonary pressures will be significantly less than systemic by three weeks of age.
All of the data obtained during the comprehensive prenatal evaluation is integrated into a prognostic profile for the baby, which predicts the likely percent survival, the need for ECMO support, the severity of pulmonary hypertension and duration of NICU stay. The CDH Team synthesizes these data in order to outline a plan for the remainder of the pregnancy, the delivery, and postnatal management.
In those fetuses with left sided CDH with an O/E LHR of <30% or right sided CDH with O/E LHR <45% may be candidates to be treated in utero with fetoscopic endoluminal balloon tracheal occlusion or FETO. FETO has been shown to significantly improve the survival of fetuses with severe left CDH by accelerating lung growth.

The baby is admitted to the NICU on the Neonatology service with the CDH Team consulting. While the Neontalogy attending will rotate during the hospitalization, the CDH Team remains consistent. A member the CDH team is on call 24/7 to address any issues with the baby. A member of the CDH Team rounds every morning with the primary team and guides the management of the baby with CDH. The CDH team also provides long-term follow-up care for all CDH babies. Parents are encouraged to join the team on morning rounds to hear updates and have any questions they may have addressed.