What is a Cephalocele?
A cephalocele is a rare condition where part of the brain or its covering pushes through an opening in the skull. These openings may be present from birth (congenital) or develop later in life (acquired).
There are two main types of cephaloceles:
- Meningoceles: These occur when the protective layers around the brain and the fluid that surrounds it, known as cerebrospinal fluid (CSF), push through an opening in the skull.
- Encephaloceles: These occur when the protective layers, CSF, and part of the brain tissue push through an opening in the skull.
Encephaloceles are classified based on where they occur on the skull. The two main locations are the posterior (back) and anterior (front) regions. Posterior encephaloceles can be further be classified into:
• Occipital: at the back of the head
• Occipitocervical: where the skull meets the neck
• Parietal: on the top side of the head
Our Care Team
At Connecticut Children’s, we understand how challenging these conditions can be for families. That’s why we have a dedicated multidisciplinary team to care for each patient. Our team is composed of specialists in Neurosurgery, Fetal Surgery, Maternal Fetal Medicine, Neonatology, Pediatric Surgery, and Fetal Radiology, who work together to create a personalized treatment plan. Our goal is to ensure that every patient receives the best possible care and support at every stage of their journey.
For Medical Professionals
Encephaloceles comprise 5% of all severe structural defects of the central nervous system that are detected in utero, and approximately 80% are detected during the first trimester (13-15). Maternal serum alpha-fetoprotein levels are of unclear significance, as encephaloceles are typically skin-covered. Therefore, prenatal imaging is the mainstay of the diagnostic workup.
Sonographic Imaging
Prenatal ultrasound has been used to detect fetal encephaloceles since the 1970s (16). Ultrasound can be used to characterize the size of the cephalocele sac, the composition of the herniated contents (to differentiate meningoceles from encephaloceles), and the size of the underlying bony defect (17, 18). Ultrasound can also be used to determine whether hydrocephalus is present, as well as to identify other abnormalities such as microcephaly, a lemon configuration of the skull, flattening of the basiocciput, or a beaked tectum (17, 19). The kidneys should be assessed, as polycystic kidneys raise the likelihood of Meckel-Gruber syndrome (an autosomal recessive syndrome characterized by the triad of encephalocele, polycystic kidneys, and polydactyly). The spine should be scanned, as well, to assess the presence of concurrent spinal dysraphism (18).
The differential diagnosis of a mass that is adjacent to the fetal cranium includes teratoma, cystic hygroma, iniencephaly, hemangioma, and a branchial cleft cyst. Several sonographic features are used to differentiate encephaloceles from other lesions. The mass must be attached to the fetal head, or at a minimum must move with the fetal head, and should form an acute angle with the surface of the fetal head (because it protrudes through a skull defect, in contrast to a cystic hygroma, which forms an obtuse angle because it arises beneath the surface of the skin) (17, 19). Identification of a skull defect is also helpful in differentiating encephaloceles from other diagnostic possibilities, but is not always possible to visualize when the skull defect is small.
Prenatal ultrasound can often be used to detect a fetal encephalocele during the first trimester (20, 21). However, the mother’s body habitus and the position of the fetus can present challenges. In such cases, endovaginal sonography may be useful (17). Additionally, cases that are impacted by Meckel-Gruber syndrome are often associated with severe oligohydramnios, which can limit the ability to perform a detailed anatomic evaluation with ultrasound during the second trimester. In such cases, close examination of the kidneys may facilitate the diagnosis during the first trimester, while there is still an adequate volume of amniotic fluid surrounding the fetus (17, 20, 22). An early diagnosis facilitates prenatal counselling and decision-making.
MRI Imaging
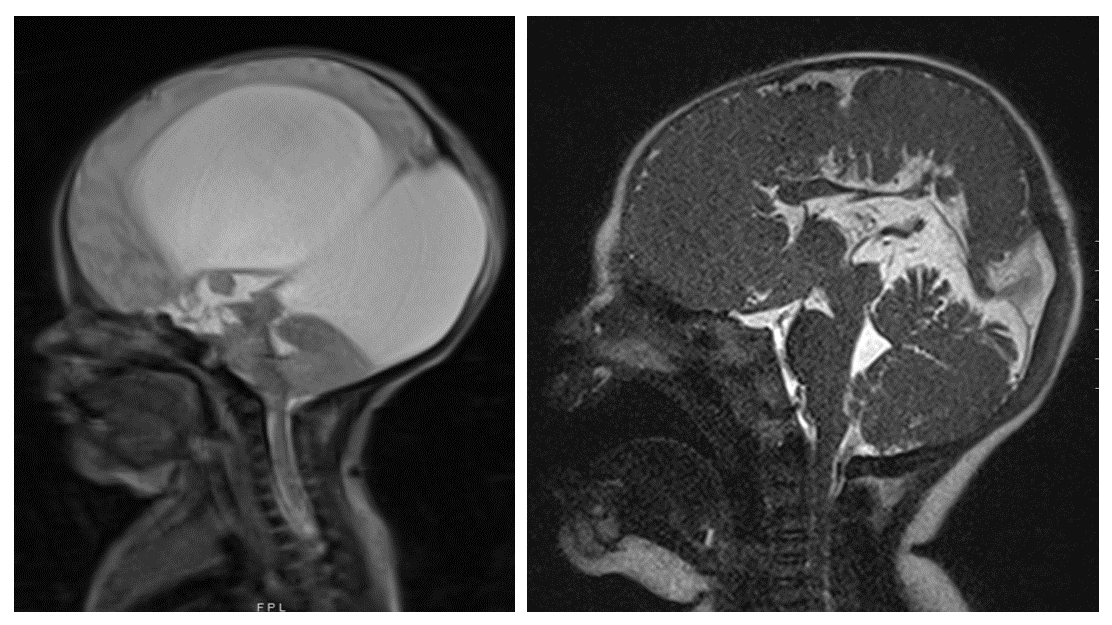
Although prenatal ultrasound is an excellent tool, fetal magnetic resonance imaging (MRI) is often used as an adjunct to obtain a more complete characterization of an encephalocele. Fetal MRI may be particularly useful when the sonographic views are limited by maternal body habitus, fetal position, or oligohydramnios. In addition to confirming the presence of a skull defect, the volume and gyral pattern of the herniated brain parenchyma can be evaluated (Figure 1) (8, 13, 23). Other cerebral malformations can also be better assessed with fetal MRI, and the spine can be evaluated for the presence of a spinal dysraphism and/or syringomyelia.
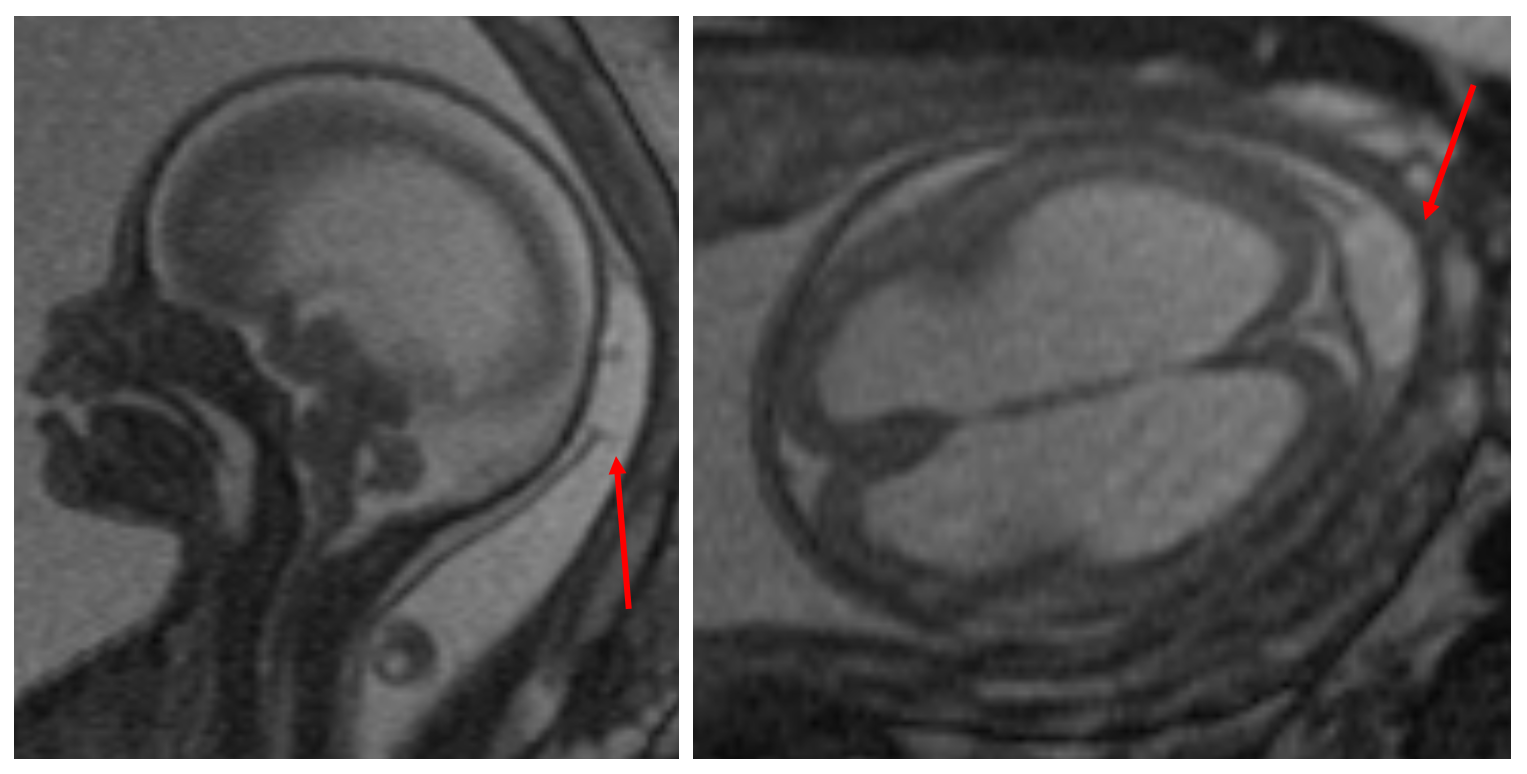
A postnatal MRI should be obtained to fully evaluate the cranial defect and characterize the herniated brain parenchyma, if any, that is present in the sac (Figure 2). MRI can also be used to identify other intracranial anomalies, such as ventriculomegaly, a Chiari malformation, Dandy-Walker malformation, holoprosencephaly, or agenesis of the corpus callosum. In addition to standard MRI sequences, vascular imaging (i.e. magnetic resonance angiography and venography) should be obtained to determine the presence of vascular structures within the sac, as well as to characterize the relationship between the cephalocele and the dural venous sinuses (8).
Surgical repair is the primary treatment for an encephalocele. The goals of surgery are to remove non-functional neural tissue, preserve functional tissue, obtain a watertight dural closure, reconstruct the bony defect (when it is large), and remove excess skin (4, 8). The surgical repair of an occipital encephalocele can typically be performed electively after several months of age, unless the skin is thin, ulcerated, and at risk of leaking CSF, in which case urgent surgery should be performed to prevent meningitis. The presence of hydrocephalus and the impact of the size of the sac on the ability to position and care for the neonate are other considerations that may impact the timing of surgery. For sincipital and basal encephaloceles, too, the risks of blood loss must be balanced with the risks of progressive facial deformity, airway compromise, and infection. In the absence of rhinorrhea, surgery is often delayed until 2-3 years of age, though some have recommended early surgery if there is sufficient nasal expansion (32).
The patient with an occipital encephalocele must be carefully intubated, either in the supine position on a donut or horseshoe head holder, or in a lateral position. The patient is then placed in the prone position, taking care to pad all bony prominences and avoid pressure on the eyes (Figure 3).
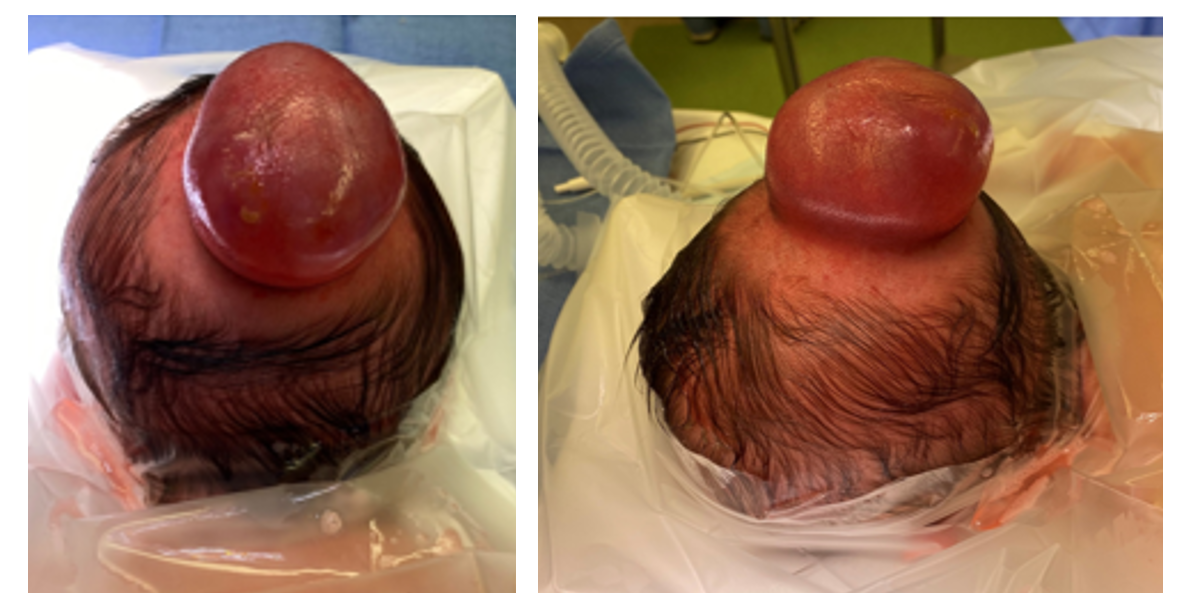
After the occipital region is sterilely prepped and draped, and preoperative antibiotics are administered, either a linear or elliptical incision is made in the skin of the encephalocele sac. The dissection is carried down to the pericranium and the bony edges are defined. Non-functional neural tissue is then coagulated and resected, while tissue that is suspected to be functional is preserved and restored to the intracranial space. Excess dura is excised, and a primary, watertight dural closure is performed (Figure 4). When a large amount of functional tissue is protruding into the cephalocele sac, the dura is closed around the herniated tissue and a cranial vault expansion can be performed, either in the same setting or a second stage procedure, to create space for the neural tissue. Autologous split thickness calvarial grafts can be used to reconstruct the bony defect when it is large; alternatively, small defects will often heal spontaneously (33).
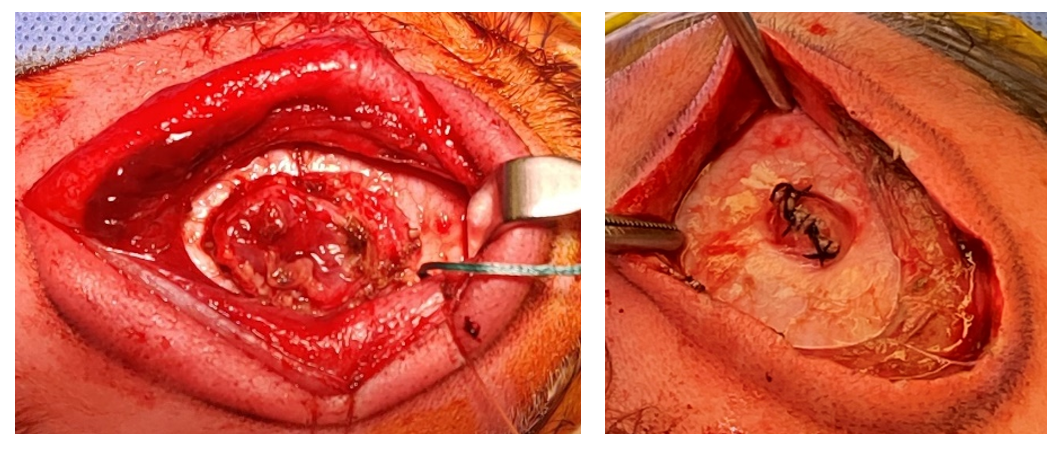
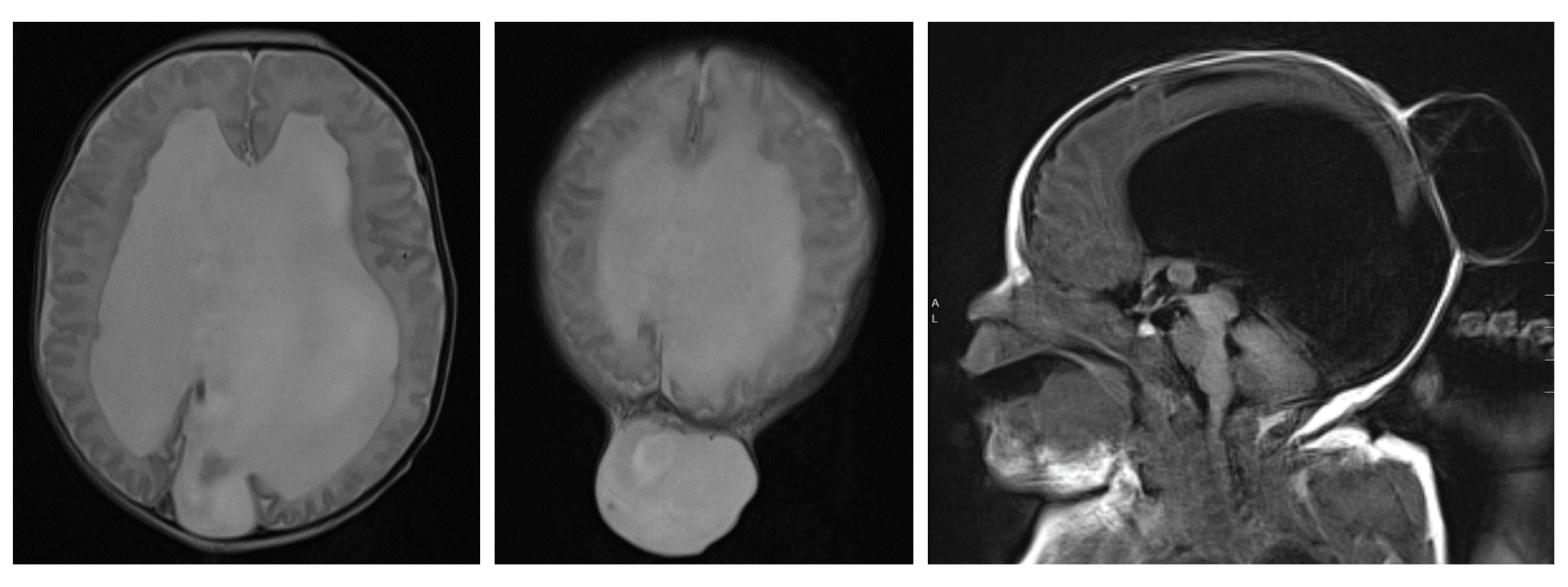
When hydrocephalus is present preoperatively, shunt placement is often performed either
before or during the encephalocele repair. Alternatively, an external ventricular drain may be placed at the time of the initial repair. When CSF diversion is not performed, close postoperative monitoring is mandatory to assess for the development of worsening hydrocephalus postoperatively. The head circumference should be tracked, serial imaging should be performed to evaluate the size of the ventricles, and the incision should be monitored for pseudomeningocele formation or CSF leakage. There should be a low threshold for shunt placement when indicated (Figure 5) (33). Endoscopic third ventriculostomy has also been described as a treatment option for encephalocele-associated hydrocephalus (34-36).