The CDH team in the Fetal Care Center at Connecticut Children’s has achieved some of the highest survival rates for congenital diaphragmatic hernia (CDH) with overall survival over 90%. There are many reasons for this exceptional survival, but the most important is the dedicated team of highly experienced neonatologists (Jeffrey Shenberger, MD, Annmarie Golioto MD) and surgeons (Timothy Crombleholme MD, Christine Finck MD) who constitute the CDH Team and manage all babies diagnosed with CDH at Connecticut Children’s Medical Center. This has allowed protocolized care, with aggressive treatment of pulmonary hypertension, integrated use of fetal echocardiogram and cardiac catheterization, very delayed surgical repair based on objective echocardiographic criteria and aggressive nutritional support to enhance pulmonary vascular remodeling and compensatory lung growth.
There is still a percentage of patients however, who do not survive despite optimal postnatal support whose pulmonary pathology may be incompatible with life. It is in these babies that the Fetal Care Center is offering fetoscopic balloon tracheal occlusion to accelerate lung growth (fig. 1).
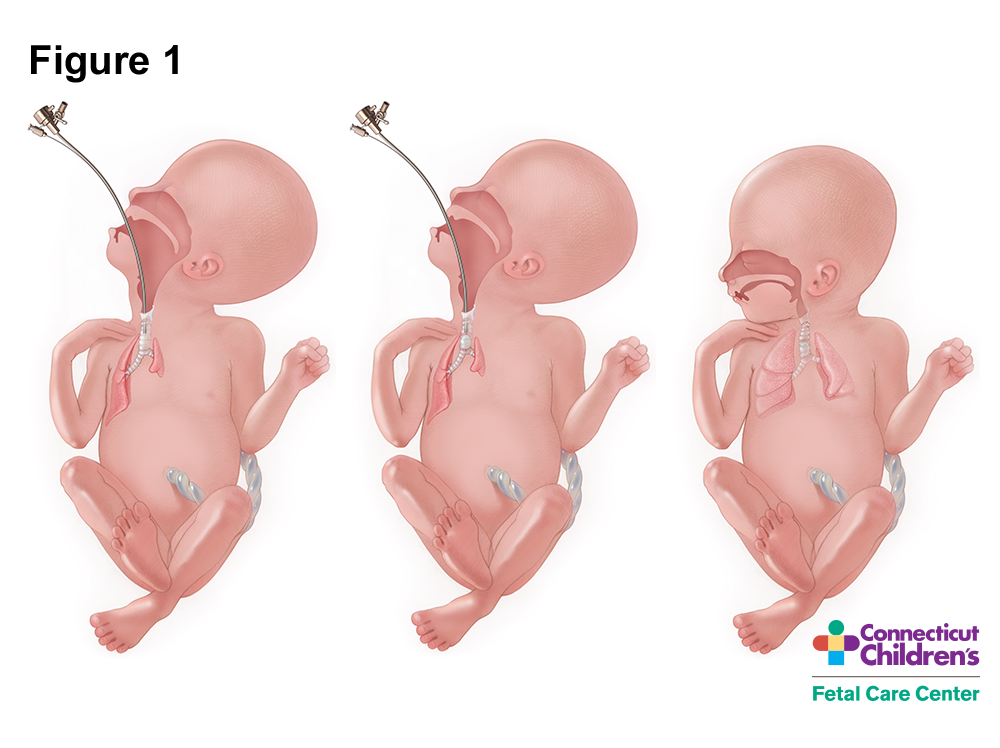
About the FETO Procedure
The critical defect in CDH is not the hole in the diaphragm or the herniated viscera (liver and intestines) but the pulmonary hypoplasia or underdevelopment of the lungs. In the womb the fetus has no need for the lungs as the placenta supplies oxygen, clears CO₂ and provides nutrients. At birth, however, the lungs must function to provide oxygen and clear CO₂ to support the baby entirely on its own. The lungs have a tremendous capacity for growth after birth, but in the setting of CDH, the lungs are hypoplastic (or underdeveloped), and may not be sufficient to provide adequate exchange of oxygen and CO₂, and because the pulmonary vasculature is similarly underdeveloped, there can be profound pulmonary hypertension (high blood pressure in the lungs). In severe CDH babies all the NICU support we can provide may still not be sufficient to help them survive. In order to improve the prognosis for these most severely affected cases of CDH, one would need to intervene before birth to accelerate both lung growth and pulmonary vascular development in order to help them survive postnatally.
The focus then shifted to occluding the fetal wind pipe or trachea to accelerate lung growth. The lungs produce fluid which the fetus breathes out into the amniotic fluid. If the fetal trachea is blocked the lung fluid builds up raising the distending pressure within the trachea and lungs stimulating accelerated lung growth. This was first done by open fetal surgery to dissect out the fetal trachea and place surgical clips across the trachea to obstruct it. This resulted in accelerated lung growth but was quite invasive increasing risks for both mother and babies. More recently, tracheal occlusion has been achieved fetoscopically by the use of a detachable balloon with fewer maternal and fetal risks.
The growth of fetal lungs is a complex process driven by natural pressure changes within the fetal airways. During the gestation the lungs do not function for gas exchange but do grow and develop. The lung continually produces fluid which is secreted into the airways. The fluid within the airways accumulates raising the intra-tracheal pressure until the natural resistance of the vocal cords and epiglottis is overcome. The fluid accumulation increases pressure within the airways and lungs stretching them a signal driving lung growth. The fetus also makes breathing movements moving fluid in and out of the lungs. When the fetus “breathes” the vocal cords and epiglottis open allowing fluids to escape from the lungs into the amniotic cavity because of the higher pressure in the lungs. The cycle of fluid accumulation leading to increased pressure triggering breathing movements and pressure release creates essential cyclical pressure changes in the lungs responsible for normal growth and development. The episodes of increased pressure causing stretch of the lungs is the primary signal for fetal lung growth and development.
An experiment of nature called Congenital High Airway Obstruction Syndrome (CHAOS) in which there is complete obstruction of the wind pipe results in enormous lungs. The obstruction prevents fluid from exiting the lungs increasing pressure which causes an increase in tissue stretch and continuously accelerated lung growth. This led to the concept of using occlusion of the fetal trachea before birth to accelerate lung growth and reverse the occlusion before birth to allow the lungs to breathe after birth. This form of fetal intervention is referred to as tracheal occlusion.
The concept of tracheal occlusion as a mechanism to accelerate lung growth was first tested and confirmed in laboratory animal experiments. These studies showed that lung growth depended on complete occlusion of the trachea, the gestational age at which it was performed and the duration of time the trachea was occluded. Tracheal occlusion also had an important adverse effect in that it decreased the development of the type of cells that make surfactant essential for lung expansion by reducing surface tension in the alveoli, the airspaces of the lung where gas exchange occurs. These special cells are called type II pneumocytes. Release of the TO allows the number of type II pneumocytes to rebound so that there is adequate surfactant production when the baby is born allowing the lungs to expand normally.
Open fetal surgery provided proof of concept that tracheal occlusion could accelerate lung growth. To avoid the risk of open fetal surgery a fetoscopic approach was developed to place a detachable balloon in the fetal airway. This approach is called Fetoscopic Endoluminal Tracheal Occlusion (FETO) and has been performed by multiple laboratories in fetal sheep confirming its effects and in hundreds of patients with improved survival compared to conventional postnatal therapy in the most severe cases of CDH.
In preliminary studies performed in the European centers Leuven, Belgium, London, United Kingdom, and Barcelona, Spain demonstrated that FETO improved survival in the most severely affected cases of left sided CDH. In a series of 210 cases of severe left CDH, FETO resulted in a 49% survival as compared to contemporaneous conventional treatment survival of only 20%. Based on these preliminary data the TOTAL trial was conducted as a prospective randomized clinical trial comparing FETO to conventional postnatal treatment. The trial had two arms: Severe and Moderate. In the severe arm, entry criteria included Observered/Expected Lung-Head Circumference ratio (O/E LHR) of < 25% and the primary endpoint of the study was survival. In the Severe arm the survival was 40%, but in the conventional postnatal treatment arm the survival was 15% (p<0.009). In the Moderate arm of the trial, the criteria for entry included O/E LHR >25< 35 + liver herniation or O/E LHR >35 <45 with liver herniation. Here too, there was a trend toward improved survival with FETO as compared to conventional postnatal treatment: 63% vs 50% (p<0.06). Due to the favorable results of the TOTAL trial the Fetal Care Center is offering FETO for left and right CDH under an FDA approved Investigational Device Exemption (IDE) and an Institutional Review Board (IRB) approved protocol.
Patients considering prenatal treatment for CDH must be evaluated at a center with FDA approval of IDE (Investigational Device Exemption) for the detachable balloon and fetoscope for its insertion that is experienced in assessing the severity if CDH using prenatal ultrasound and fetal MRI. This evaluation includes excluding other potentially associated anomalies, placental location and fetal position. Prognostic indicators are measured including the lung-head circumference ratio, or LHR, as well as the observed/expected LHR (O/E LHR), modified McGoon index to help predict the severity of pulmonary hypertension. Fetal MRI is also used to define severity of lung herniation, total lung volume (TLV) and percent predicted lung volume (PPLV).
A multidisciplinary team is used to evaluate and counsel potential candidates for FETO including Maternal Fetal Medicine Specialists, Fetal and Pediatric Surgeons, Neonatologists, Radiologists, Medical Social Worker, and members of the CDH Team.
The fetus must have isolated left sided diaphragmatic hernia with or without herniation of the liver. The O/E LHR must be ≤30% to qualify for the procedure. In the case of right sided CDH the O/E LHR must be <45%. A normal full karyotype analysis or at least a normal FISH must be obtained to qualify. Multiple other criteria will be measured, such as the TLV, PPLV and modified McGoon index to stratify risk but these are not criteria for FETO.
Patients are asked to eat and drink nothing after midnight and are admitted just prior to the surgery. An ultrasound is performed to check the fetal position, a toco-dynomometer is placed to record uterine activity. A written consent must be obtained prior to surgery.
Fetsocopic surgery is most commonly performed under an epidural anesthetic and rarely is general anesthesia required. Local anesthesia is possible in highly selected cases. The fetus also receives medication so that it does not feel discomfort and does not move during the operation. Occasionally, the fetal position must be adjusted by external manipulation to facilitate fetoscopic entry into the mouth and trachea.
After sterilely prepping and draping, a 4 mm incision is made in the maternal skin to introduce a curved 3.3 mm sheath into the amniotic cavity under ultrasound guidance (fig. 2). A small fetoscope with attached camera is placed through the sheath and surgeon views what is in the amniotic cavity on the video screen. The fetal surgeon maneuvers the fetoscope into the fetal mouth, under the epiglottis and through the vocal cords into the fetal trachea (windpipe). Just above the bifurcation of the windpipe into the right and left bronchi a small balloon is inserted, inflated and detached. Antibiotics are delivered into the amniotic fluid before the fetoscope is removed and the small skin incision is closed with a single stitch. The mother is monitored overnight and may get medication to keep the uterus from contracting. Most mothers will be allowed to leave the hospital on postoperative day 1.
The day after the procedure and at least weekly thereafter an ultrasound will be obtained to measure the position of the balloon and the LHR and O/E LHR. Because the baby’s trachea is obstructed by the balloon it is essential that mothers remain in close proximity to the Fetal Care Center, either nearby hotel or at a family or friend’s house within 20 minute drive to the Fetal Care Center.
If for whatever reason the baby is born, the balloon must be removed to allow the baby to breathe. Removal of the balloon can be performed at the time of delivery, but ideally should be done before delivery. Earlier removal allows time for the number of type II pneumocytes to recover and adequate surfactant to be made.
Although an unplanned delivery is unlikely, the baby may come earlier than planned. The mother must be able to travel to the Fetal Care Center where a team is equipped and experienced in techniques for balloon removal and in case of an emergency is available 24/7. Patients not willing to stay near the Fetal Care Center are not candidates for FETO as an early delivery can create and emergency situation which might not be able to be handled by local caregivers as they are not familiar with the procedure.
If the mother experiences fluid leakage, she most likely has ruptured membranes, she needs to be examined at the Fetal Care Center and if confirmed will be admitted for inpatient management. In most cases this does not lead immediately to contractions, but the risks for it are increased. If contractions occur and labor cannot be arrested, everything will be prepared for balloon removal in the most appropriate method: fetoscopic retrieval, ultrasound guided needle puncture, or Ex-utero Intrapartum Treatment (EXIT) procedure. The balloon can even be popped by passing a specially designed needle through the endotracheal tube after delivery.
At 34 weeks the mother will be readmitted to the Fetal Care Center. Medications are administered to stimulate lung maturity and surfactant production (steroids), antibiotics to prevent infection and medications to prevent uterine contractions. The balloon can be retrieved fetoscopically just as it was inserted. Under direct fetoscopic vision the tail of the balloon is grasped, punctured and removed. Alternatively, the balloon can be needle punctured under ultrasound guidance. The balloon remnant is left to be coughed out by the fetus into the amniotic cavity.
After balloon removal the mother is monitored in the hospital overnight with an ultrasound the next day and discharged from the hospital. The pregnancy is continued and delivery is normally planned at 38 weeks at the Fetal Care Center.
Rarely, it is necessary to remove the balloon via an EXIT procedure which is a modified Cesarean Section during which the baby is partially delivered still on placental support while bronchoscopy (scope passed into trachea) is performed to remove the balloon. If such a procedure needs to be performed, the baby will be born at the time of balloon removal.
Babies with CDH are ideally born in optimal conditions planned ahead of time in order to have all required team members available. If there are no problems with the pregnancy this will be planned for ~38 weeks. The immediate postnatal care of CDH babies consists of resuscitation and stabilization, respiratory support via conventional mechanical ventilation or high frequency oscillatory ventilation, the use of inhaled nitric oxide, steroids and agents to maintain blood pressure. Surgical repair is deferred until estimated pulmonary pressures are less than 80% of systemic as indicated by echocardiography.
The cumulative experience with FETO worldwide is now well over 300 procedures. The results of the TOTAL trial show that, in cases of severe pulmonary hypoplasia (O/E LHR ≤25) with anticipated postnatal survival with conventional therapy of only 15-20%, FETO has a 40% survival rate. In these cases balloon insertion was between 26-28 weeks with scheduled removal at 34 weeks. The average operation time was 10-15 minutes. In ~3% of cases it was not possible to place the balloon in the correct position or the first attempt mainly due to difficult position of the baby or deflation of the balloon requiring a second balloon.
The results of the North American FETO Consortium have observed improved survival with both FETO (60%) and postnatal conventional treatment (40%) compared to the TOTAL trial results. However, these observation are from a retrospective/prospective cohort study from 9 institutions and not a prospective randomized trial. The differences in survival did not achieve statistical significance.
Overall there appears to be a 30-35% improvement in survival compared to what can be expected with postnatal management (49% vs. 25%). But these studies were not randomized. The response of lung growth is proportional to the size of the lung prior to the procedure. That is, the fetuses with larger lungs among the fetuses with the O/E LHR ≤25% respond better than those fetuses with extremely small lungs in the same category.
Fetoscopic surgery is a minimally invasive procedure with which there is extensive experience in fetal surgery and there are no known serious complications for the mother.
The most common and most important complication with FETO is early rupture of membranes with vaginal leakage of amniotic fluid. This problem occurs within one week of FETO in about 5% of cases and within 6 weeks in 20% of cases. The leakage of amniotic fluid may continue for several weeks until delivery. In some cases the leakage stops and the pregnancy continues normally. Unfortunately, in other cases this rupture of membranes leads to preterm labor and delivery. The consequences of preterm birth depend on the gestational age at which this occurs. Normally, babies born after 32 weeks usually survive without any long term problems, but may require neonatal intensive care. However, prematurity in CDH may increase the risks of deaths or long-term breathing and feeding problems.
In animal experiments and in most human experience with FETO the balloon causes no damage to the trachea. In a few babies however, there is local widening of the trachea. It is not clear if this was due to the balloon, the postnatal endotracheal tube or other complications arising from CDH. We assume this widening is due to the balloon and has resulted in mild breathing problems in a few cases, specifically tracheomalacia associated with a barking cough.
Minor other complications that could occur in more than 1% of cases exclude localized bleeding or wound infection at the entry site of the instruments.